19 stable releases
| 2.0.3 | Mar 26, 2025 |
|---|---|
| 2.0.1 | Jan 24, 2025 |
| 1.1.9 | Oct 30, 2024 |
| 1.0.6 | Jul 24, 2024 |
#17 in Biology
247 downloads per month
1MB
7.5K
SLoC
Proseg
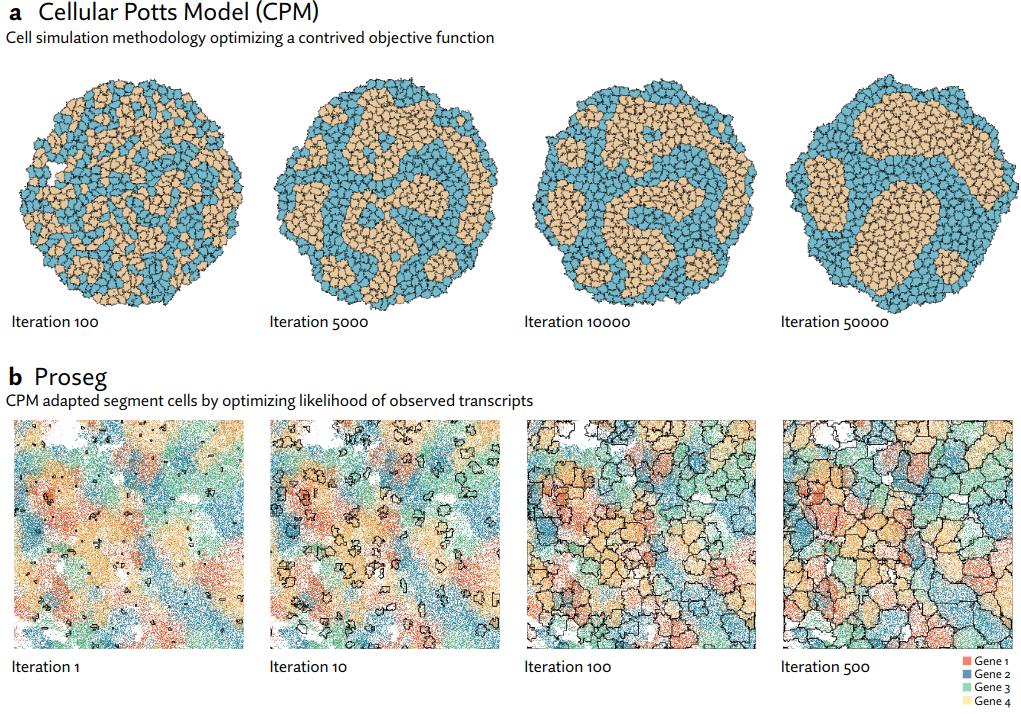
Proseg (probabilistic segmentation) is a cell segmentation method for in situ spatial transcriptomics. Xenium, CosMx, and MERSCOPE platforms are currently supported.
Installing
Proseg can be built and installed with cargo. Clone this repository, then run
cargo install proseg
General usage
Proseg is run on a table of transcript positions which in some form must include preliminary assignments of transcripts to nuclei. Xenium, CosMx, and MERSCOPE all provide this out of the box in some form.
Proseg is invoked, at minimum like:
proseg /path/to/transcripts.csv.gz
There are command line arguments to tell it which columns in the csv file to use,
but typically one of the presets --xenium, --cosmx, or --merfish are used.
Proseg is a sampling method, and in its current form in non-deterministic. From run to run, results will vary slightly.
General options
By default proseg will use all available CPU cores. To change this use --nthreads N.
Output options
Output is in the form of a number of tables, which can be either gzipped csv files or parquet files, and GeoJSON files giving cell boundaries.
--output-expected-counts expected-counts.csv.gz: Cell-by-gene count matrix. Proseg is a sampling method, so these are posterior expectations that will generally not be integers but fractional counts.--output-cell-metadata cell-metadata.csv.gz: Cell centroids, volume, and other information.--output-transcript-metadata transcript-metadata.csv.gz: Transcript ids, genes, revised positions, assignment probability, etc.--output-gene-metadata: Per-gene summary statistics--output-rates rates.csv.gz: Cell-by-gene Poisson rate parameters. These are essentially expected relative expression values, but may be too overly-smoothed for use in downstream analysis.
Cell boundaries can be output a number of ways:
--output-cell-polygons cell-polygons.geojson.gz: 2D polygons for each cell in GeoJSON format. These are flattened from 3D, so will overlap.--output-cell-polygon-layers cell-polygons-layers.geojson.gz: Output a separate, non-overlapping cell polygon for each z-layer, preserving 3D segmentation.--output-cell-hulls cell-hulls.geojson.gz: Instead of inferred cell polygons, output convex hulls around assigned transcripts.--output-cell-voxels cell-voxels.csv.gz: Output a (very large) table giving the coordinates and cell assignment of every assigned voxel.
Modeling assumptions
A number of options can alter assumptions made by the model, which generally should not need
--ncomponents 5: Cell gene expression is a modeled as a mixture of negative binomial distributions. This parameter controls the number of mixture components. More components will tend to nudge the cells into more distinct types, but setting it too high risks manifesting cell types that are not real.--no-diffusion: By default Proseg models cells as leaky, under the assumption that some amount of RNA leaks from cells and diffuses elsewhere. This seems to be the case in much of the Xenium data we've seen, but could be a harmfully incorrect assumption in some data. This argument disables that part of the model.--diffusion-probability: Prior probability of a transcript is diffused and should be repositioned.--diffusion-sigma-far: Prior standard deviation on transcript repositioning distance.--voxel-layers 4: Number of layers of voxels on the z-axis to use. Essentially how 3D the segmentation should be.--initial-voxel-size 4: Initial side length of voxels on the xy-axis.--schedule 150,150,300: A comma separated list of numbers giving the sampling schedule. The sampler runs for a given number of iterations, halves the voxel size, then runs for the next number of iterations.--nuclear-reassignment_prob 0.2: Prior probability that the initial nuclear assignment (if any) is incorrect.--perimeter-bound 1.3: Larger numbers allow less spherical cells.
General advice
- If the dataset conisists of multiple tissue sections, it's generally safer to split these and segment them separately. This avoids potential sub-optimal modeling due to different z-coordinate distributions.
Running on Xenium datasets
Xenium data should be run with the --xenium argument.
Using Xenium Explorer with proseg-to-baysor
It is possible to use proseg segmentation with Xenium Explorer, but requires a little work.
The xeniumranger tool has a command to import segmentation from Baysor. To use this, we must first convert Proseg output to Baysor-compatible formatting.
For this we need transcript metadata and cell polygons from Proseg, then run the provided proseg-to-baysor
command like
proseg-to-baysor \
transcript-metadata.csv.gz \
cell-polygons.geojson.gz \
--output-transcript-metadata baysor-transcript-metadata.csv \
--output-cell-polygons baysor-cell-polygons.geojson
Xenium Ranger can then be run to import these into a format useable with Xenium Explorer:
xeniumranger import-segmentation \
--id project-id \
--xenium-bundle /path/to/original/xenium/output \
--viz-polygons baysor-cell-polygons.geojson \
--transcript-assignment baysor-transcript-metadata.csv \
--units microns
This will output a new Xenium bundle under the project-id directory
Xenium Explorer currently has issues displaying Proseg polygons. It appears to
perform some sort of naive polygon simplification that results in profoundly
distorted polygons. There's not any known workaround for this issue for now.
Issues displaying proseg polygons in Xenium Explorer are resolved with more recent versions of Xenium Ranger (starting with 2.0).
Running on CosMx datasets
Earlier versions of CosMx did not automatically provide a single table of global
transcript positions. To work around this, we provide a Julia program in
extra/stitch-cosmx.jl to construct a table from the flat files downloaded from
AtoMx.
To run this, some dependencies are required, which can be installed with
julia -e 'import Pkg; Pkg.add(["Glob", "CSV", "DataFrames", "CodecZlib", "ArgParse"])'
Then the program can be run with like
julia stitch-cosmx.jl /path/to/cosmx-flatfiles transcripts.csv.gz
to output a complete transcripts table to transcripts.csv.gz.
From here proseg can be run with
proseg --cosmx-micron transcripts.csv.gz
Alternatively, the --cosmx can be used with CosMx data that is in pixel coordinates.
It will automatically scale the data to micrometers.
Running on MERSCOPE datasets
No special considerations are needed for MERSCOPE data. Simply use the
--merscope argument with the detected_transcripts.csv.gz file.
Dependencies
~47–75MB
~1.5M SLoC