11 releases
| 0.5.1 | Jun 23, 2024 |
|---|---|
| 0.5.0 | Aug 5, 2023 |
| 0.4.0 | May 4, 2023 |
| 0.3.0 | Nov 16, 2022 |
| 0.1.4 | Jul 7, 2021 |
#21 in Biology
91 downloads per month
445KB
6.5K
SLoC
block aligner
![]()
![]()
SIMD-accelerated library for computing global and X-drop affine gap penalty sequence-to-sequence or sequence-to-profile alignments using an adaptive block-based algorithm.
See the Bioinformatics paper here for more info on the algorithm and how it compares with other algorithms.
If you found Block Aligner useful, please cite:
@article{liu2023block,
title={{Block Aligner}: an adaptive {SIMD}-accelerated aligner for sequences and position-specific scoring matrices},
author={Liu, Daniel and Steinegger, Martin},
journal={Bioinformatics},
volume={39},
number={8},
pages={btad487},
year={2023},
publisher={Oxford University Press}
}
Example
use block_aligner::{cigar::*, scan_block::*, scores::*};
let min_block_size = 32;
let max_block_size = 256;
// A gap of length n will cost: open + extend * (n - 1)
let gaps = Gaps { open: -2, extend: -1 };
// Note that PaddedBytes, Block, and Cigar can be initialized with sequence length
// and block size upper bounds and be reused later for shorter sequences, to avoid
// repeated allocations.
let r = PaddedBytes::from_bytes::<NucMatrix>(b"TTAAAAAAATTTTTTTTTTTT", max_block_size);
let q = PaddedBytes::from_bytes::<NucMatrix>(b"TTTTTTTTAAAAAAATTTTTTTTT", max_block_size);
// Align with traceback, but no X-drop threshold (global alignment).
let mut a = Block::<true, false>::new(q.len(), r.len(), max_block_size);
a.align(&q, &r, &NW1, gaps, min_block_size..=max_block_size, 0);
let res = a.res();
assert_eq!(res, AlignResult { score: 7, query_idx: 24, reference_idx: 21 });
let mut cigar = Cigar::new(res.query_idx, res.reference_idx);
// Compute traceback and resolve =/X (matches/mismatches).
a.trace().cigar_eq(&q, &r, res.query_idx, res.reference_idx, &mut cigar);
assert_eq!(cigar.to_string(), "2=6I16=3D");
See the docs for detailed API information.
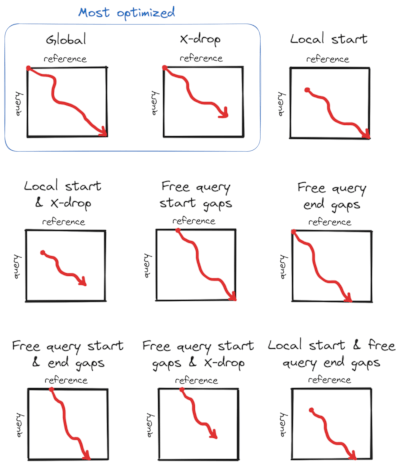
Here is a diagram showing alignment types that are supported in Block Aligner:
Algorithm
Block aligner provides a new efficient way to compute pairwise alignments on proteins, DNA sequences, and byte strings with dynamic programming. Block aligner also supports aligning sequences to profiles, which are position-specific scoring matrices and position-specific gap open costs.
It works by calculating scores in a small square block that is shifted down or right in a greedy manner, based on the scores at the edges of the block. This dynamic approach results in a much smaller calculated block area compared to previous approaches, though at the expense of some accuracy. The block can also go back to a previous best checkpoint and grow larger, to handle difficult regions with large gaps. The block size can also dynamically shrink when it detects that a large block is not needed. Both block growing and shrinking are based on heuristics.
By trading off some accuracy for speed, block aligner is able to efficiently handle a variety of scoring matrices and adapt to sequences of varying sequence identities. In practice, it is still very accurate on a variety of protein and nucleotide sequences.
Block aligner is designed to exploit SIMD parallelism on modern CPUs. Currently, SSE2 (128-bit vectors), AVX2 (256-bit vectors), Neon (128-bit vectors), and WASM SIMD (128-bit vectors) are supported. For score calculations, 16-bit score values (lanes) and 32-bit per block offsets are used.
Block aligner behaves similarly to an (adaptive) banded aligner when the minimum and maximum block size is set to the same value.
Tuning block sizes
For long, noisy Nanopore reads, a min block size of ~1% sequence length and a max block size
of ~10% sequence length performs well (tested with reads up to ~50kbps).
For proteins, a min block size of 32 and a max block size of 256 performs well.
Using a minimum block size that is at least 32 is recommended for most applications.
Using a maximum block size greater than 2^14 = 16384 is not recommended.
The library contains a percent_len function that computes a percentage of the sequence length with these recommendations.
If the alignment scores are saturating (score too large), then use a smaller block size.
Let me know how block aligner performs on your data!
Install
This library can be used on both stable and nightly Rust channels. The nightly channel is needed for running tests and benchmarks. Additionally, the tests and benchmarks need to run on Linux or MacOS.
To use this as a crate in your Rust project, add the following to your Cargo.toml:
[dependencies]
block-aligner = { version = "0.5", features = ["simd_avx2"] }
Use the simd_sse2, simd_neon, or simd_wasm feature flag for x86 SSE2, ARM Neon, or WASM SIMD support, respectively.
It is your responsibility to ensure the correct feature to be enabled and supported by the
platform that runs the code because this library does not automatically detect the supported
SIMD instruction set. More information on specifying different features for different platforms
with the same dependency here.
Here's a simple example:
[target.'cfg(target_arch = "x86_64")'.dependencies]
block-aligner = { version = "0.5", features = ["simd_avx2"] }
[target.'cfg(target_arch = "aarch64")'.dependencies]
block-aligner = { version = "0.5", features = ["simd_neon"] }
For developing, testing, or using the C API, you should clone this repo and use Rust nightly. In general, when building, you need to specify the correct feature flags through the command line.
For x86 AVX2:
cargo build --features simd_avx2 --release
For x86 SSE2:
cargo build --features simd_sse2 --release
For ARM Neon:
cargo build --target=aarch64-unknown-linux-gnu --features simd_neon --release
For WASM SIMD:
cargo build --target=wasm32-wasi --features simd_wasm --release
To run WASM programs, you will need wasmtime
installed and on your $PATH.
C API
There are C bindings for block aligner. More information on how to use them is located in
the C readme.
See the 3di branch for an example of using block aligner to do local alignment in C,
along with block aligner modifications to support aligning with amino acid 3D interaction (3Di) information.
Improving Block Aligner
During alignment, three decisions need to be made at each step (using heuristics):
- Whether to grow the block size
- Whether to shrink the block size
- Whether to shift right or down
Block aligner uses simple greedy heuristics that are cheap to evaluate for making these decisions. There is probably a lot of room to improve here! Maybe seeds? Neural network models?
To try your ideas, take a look at the code after the comment // TODO: better heuristics? in src/scan_block.rs
(depending on your changes, you may need to modify other parts of the code too). Let me know if you
are working on new ideas!
Most of the instructions below are for benchmarking and testing block aligner.
Data
Some Illumina/Nanopore (DNA), Uniclust30 (protein), and SCOP (protein profile) data are used in some tests and benchmarks. You will need to download them by following the instructions in the data readme.
Test
Run scripts/test_avx2.sh or scripts/test_wasm.sh to run tests.
CI will run these tests when commits are pushed to this repo.
More testing and evaluating scripts are available in the scripts directory.
For debugging, there exists a debug feature flag that prints out a lot of
useful info about the internal state of the aligner while it runs.
There is another feature flag, debug_size, that prints the sizes of blocks after they grow.
To manually inspect alignments, run scripts/debug_avx2.sh with two sequences as arguments.
Docs
Run scripts/doc_avx2.sh or scripts/doc_wasm.sh to build the docs locally.
Benchmark
Run scripts/bench_avx2.sh or scripts/bench_wasm.sh for basic benchmarks.
See the scripts directory for runnable benchmark scripts on real data.
Most of the actual implementations of the benchmarks are in the examples directory.
Data analysis and visualizations
Use the Jupyter notebook in the vis/ directory to gather data and plot them. An easier way
to run the whole notebook is to run the vis/run_vis.sh script. This reproduces the
experiments in the manuscript.
Profiling with MacOS Instruments
Use
brew install cargo-instruments
RUSTFLAGS="-g" cargo instruments --example profile --release --features simd_avx2 --open
Analyzing performance with LLVM-MCA
Use
scripts/build_ir_asm.sh
to generate assembly output and run LLVM-MCA.
Viewing the assembly
Use either scripts/build_ir_asm.sh, objdump -d on a binary (avoids recompiling code in
some cases), or a more advanced tool like Ghidra (has a decompiler, too).
Compare (relatively unused)
Edits were made to Hajime Suzuki's adaptive banding benchmark code and difference recurrence benchmark code. These edits are available here and here, respectively. Go to those repos, then follow the instructions for installing and running the code.
If you run the scripts in those repos for comparing scores produced by different algorithms,
you should get .tsv generated files. Then, in this repo's directory, run
scripts/compare_avx2.sh /path/to/file.tsv 50
to get the comparisons. The X-drop threshold is specified after the path.
Old ideas and history
See the ideas file.